SeqCode: File formats
General description
General information on the multiple input and output file formats processed by SeqCode.
Genome Definition: chromosomes
SeqCode defines the content of the genome from one file containing the name and the size of the chromosomes of
the organism. The UCSC genome browser provides this information through the
Downloads section for each genome (Annotation database link). Thus, users can retrieve the corresponding
chromInfo.txt file for the appropriate organism. For example, the mouse file (mm9) is accessible from
http://hgdownload.soe.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz. These files must be uncompressed previously to be used with SeqCode and the name of
alternative contigs (e.g. chr13_random) can be dismissed into the final archive.
This is an example of this class of files for mouse (genome assembly: mm9):
This is an example of this class of files for mouse (genome assembly: mm9):
chr1 197195432 chr10 129993255 chr11 121843856 chr12 121257530 chr13 120284312 chr14 125194864 chr15 103494974 chr16 98319150 chr17 95272651 chr18 90772031 chr19 61342430 chr2 181748087 chr3 159599783 chr4 155630120 chr5 152537259 chr6 149517037 chr7 152524553 chr8 131738871 chr9 124076172 chrM 16299 chrX 166650296 chrY 15902555
Gene Models: RefGene format
SeqCode uses the gene transcript annotations provided by the RefSeq
consortium to define the location of genomic features on virtually any genome that is served by this project.
The UCSC genome browser provides this information through the Downloads
section for each genome (Annotation database link). Thus, users can retrieve the corresponding refGene.txt.gz
file for the appropriate organism. For example, the mouse file (mm9) is accessible from
http://hgdownload.soe.ucsc.edu/goldenPath/mm9/database/refGene.txt.gz. Such files must be uncompressed previously to be used with SeqCode (e.g. gzip -d refGene.txt.gz).
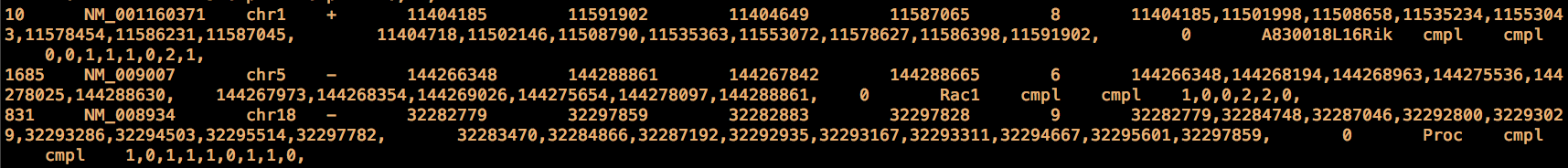
This is an extract of the refGene.txt file for mouse (genome assembly: mm9), each line contains the information for a RefSeq transcript:
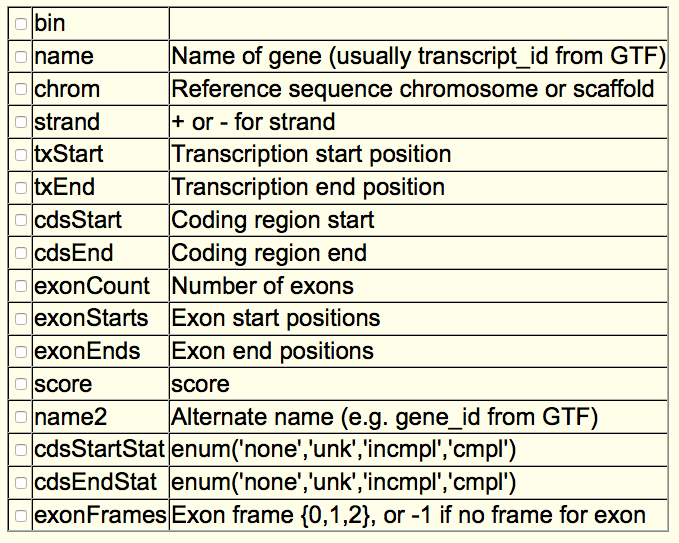
This is the description of each field in this class of files according to the UCSC table browser:
This is an extract of the refGene.txt file for mouse (genome assembly: mm9), each line contains the information for a RefSeq transcript:
This is the description of each field in this class of files according to the UCSC table browser:
High-throughput Sequencing Data: BAM/SAM format
SeqCode mainly analyzes the set of mapped reads of a sequencing experiment (e.g. ChIPseq/RNAseq/ATACseq) in
SAM format. This file format contains one line per read
that is mapped, containing the location in the genome for such a read. BAM files, which are the compressed version
of SAM files, can be also provided to SeqCode. For further information, users can access the SAM format documentation at
https://samtools.github.io/hts-specs/SAMv1.pdf.
This is an extract of a SAM file from the mapping of a ChIPseq experiment:

Implementation notes (SeqCode requirements):
- SAM/BAM files should exclusively contain aligned reads (unmapped reads with flag=4 must be removed).
- SAM/BAM file indexing and sorting is not required to perform SeqCode analyses.
This is an extract of a SAM file from the mapping of a ChIPseq experiment:
Implementation notes (SeqCode requirements):
- SAM/BAM files should exclusively contain aligned reads (unmapped reads with flag=4 must be removed).
- SAM/BAM file indexing and sorting is not required to perform SeqCode analyses.
Genome Regions: BED format
The location of ChIPseq peaks or any other genomic feature can be easily represented using the BED format
(Browser Extensible Data, see the
UCSC documentation for further information).
In the simplest version of this format, each line in these files contains 4 columns: chromosome, initial position,
ending position and (optionally) a name for identifying this element.
This is an example of a BED format file:
This is an example of a BED format file:
chr1 4481268 4484041 MACS_peak_1 chr1 4485405 4487849 MACS_peak_2 chr1 5008529 5011367 MACS_peak_3 chr1 9788194 9789005 MACS_peak_4 chr1 9838297 9839446 MACS_peak_5 (...)
Genome-wide Profiles: BedGraph format
SeqCode generates custom tracks for visualization in genome browsers using the BedGraph format. With this format,
it is possible to produce distribution functions to map the location of high-throughput experiments or other features
along the chromosomes (more information on the
UCSC genome browser documentation).
Basically, it is derived from the BED format by adding a number in the 4th column to define the height of the profile
in that region.
This is an excerpt of this type of files:
This is an excerpt of this type of files:
track type=bedGraph name="NAME" description="TEXT" visibility="full" color="0,0,100" chr1 2999917 3000116 1 chr1 3000117 3000241 1 chr1 3001026 3001226 1 chr1 3001269 3001468 1 chr1 3001469 3001580 1 chr1 3002287 3002486 1 chr1 3002487 3002566 1 chr1 3002567 3002735 2 chr1 3002736 3002767 1 chr1 3003804 3004004 1 (...)
Evolutionary sequence conservation: PhastCons scores
To calculate the conservation of a genomic region, SeqCode uses the PhastCons data files provided by the
UCSC genome browser. These files provide chromosome by chromosome the scores
of the multiple comparison between the reference species and a group of other genomes. For instance, the folder
http://hgdownload.soe.ucsc.edu/goldenPath/mm9/phastCons30way/
provides the data files for the phastCons30way track in which mouse is compared to 30 vertebrates. Please, note
that such files must be uncompressed previously to be used with SeqCode (estimated average size per mouse chromosome around
1 GigaByte).
This is an excerpt of this type of files for mouse (genome assembly: mm9):
This is an excerpt of this type of files for mouse (genome assembly: mm9):
fixedStep chrom=chr1 start=3000306 step=1 0.006 0.010 0.014 0.017 0.019 0.021 0.021 0.021 0.019 (...)