SeqCode: Program Usage
General description
SeqCode is designed to run under command-line platforms.
Command-line interfaces are running
environments where the user interacts with the computer by introducing text commands. SeqCode is able to
run under Linux and
MacOSX operating systems through the command-line interface
of the Terminal (the shell). The final behavior of such programs can be configured by adding options using
the symbol '-' plus the character defining such an option. The output of the program is usually stored into
an output folder in which the user will find the resulting plots, the scripts to generate the plots in R (if any),
and the data files.
This is the prototype of a SeqCode call from the Terminal (the final format will depend on each SeqCode program):
Each function of SeqCode is provided by an stand-alone program. This is the current list of available applications:
All SeqCode programs include the option '-h' to visualize the basic documentation and the full list of options to configure it. This is an example when using this option for the buildChIPprofile command to show the help information:
This is the prototype of a SeqCode call from the Terminal (the final format will depend on each SeqCode program):
> SeqCode_command -options input_file1 ... input_fileN OutputFolderName
Each function of SeqCode is provided by an stand-alone program. This is the current list of available applications:
buildChIPprofile combineChIPprofiles combineTSSmaps combineTSSplots computemaxsignal findPeaks genomeDistribution matchpeaks matchpeaksgenes processmacs produceGENEmaps produceGENEplots producePEAKmaps producePEAKplots produceTESmaps produceTESplots produceTSSmaps produceTSSplots recoverChIPlevels scorePhastCons
All SeqCode programs include the option '-h' to visualize the basic documentation and the full list of options to configure it. This is an example when using this option for the buildChIPprofile command to show the help information:
> buildChIPprofile -h SeqCode_v1.0 User commands buildChIPprofile NAME buildChIPprofile - a program to generate genome-wide ChIPseq/ATACseq/RNAseq profiles for genome browser visualization. SYNOPSIS buildChIPprofile [-c][-d][-l ][-m][-p][-s ][-w ][-v][-x prefix][-h] OUTPUT One folder with one file: - BedGraph profile (compressed). OPTIONS -c : Track color (default: black). -d : Demo mode for small BAM files (min number reads control off). -l : Avg. fragment size (default: 150). -m : Invert the score of BedGraph records. -p : Using pair-end reads mapped in proper pair (default: single-end). -s : Number of spike-in reads for reference-adjusted normalization. -w : Window resolution (default: 100). -v : Verbose. Display info messages. -x : Prefix for the output folder. -h : Show this help. SEE ALSO SeqCode homepage: http://ldicrocelab.crg.es GitHub source code: https://github.com/eblancoga/seqcode AUTHORS Written by Enrique Blanco. SeqCode_v1.0 User commands buildChIPprofile
How To Access SeqCode Output
Most SeqCode commands generate one folder with the resulting output once the execution is finished. In all cases,
the user must enter the name that will be used by SeqCode to generate this folder before. The precise location of
the output folder in the path can be customized with the option -x (prefix). Those commands involving
the production of PDF plots will include the final PDF file together with the R script and the data file to generate
it into this folder. This is an example of the output when running the produceTSSplots command on a sample called TEST1:
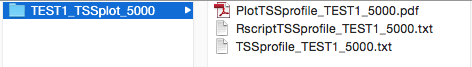
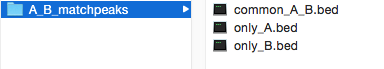
Output folders of SeqCode commands that do not generate PDF plots for visualization will only include the resulting data files (e.g. BedGraph files for custom tracks or matching/mismatching peaks for intersection of peaks). This is an example of the output of the matchpeaks command on two sets called A and B:
The matchpeaksgenes command is the only one that directly delivers the results through the standard output, allowing to easily integrate this command on a pipeline to process the target genes of the analysis (the H3K4me3 ChIPseq sample in the following example):
Output folders of SeqCode commands that do not generate PDF plots for visualization will only include the resulting data files (e.g. BedGraph files for custom tracks or matching/mismatching peaks for intersection of peaks). This is an example of the output of the matchpeaks command on two sets called A and B:
The matchpeaksgenes command is the only one that directly delivers the results through the standard output, allowing to easily integrate this command on a pipeline to process the target genes of the analysis (the H3K4me3 ChIPseq sample in the following example):
> matchpeaksgenes -u 2500 H3K4me3_mESC.bed refGene.txt | head -10
chr1 3521328 3522342 MACS_peak_1 Xkr4 NM_001011874 chr1 - 3204562 3664079
chr1 3660177 3663163 MACS_peak_2 Xkr4 NM_001011874 chr1 - 3204562 3664079
chr1 4481311 4484482 MACS_peak_4 Sox17 NM_001289464 chr1 - 4481008 4489935
chr1 4481311 4484482 MACS_peak_4 Sox17 NM_001289465 chr1 - 4481008 4489935
chr1 4481311 4484482 MACS_peak_4 Sox17 NM_001289466 chr1 - 4481008 4489935
chr1 4481311 4484482 MACS_peak_4 Sox17 NM_001289467 chr1 - 4481008 4489935
chr1 4481311 4484482 MACS_peak_4 Sox17 NM_011441 chr1 - 4481008 4489935
chr1 4486175 4488222 MACS_peak_5 Sox17 NM_001289464 chr1 - 4481008 4489935
chr1 4486175 4488222 MACS_peak_5 Sox17 NM_001289465 chr1 - 4481008 4489935
chr1 4486175 4488222 MACS_peak_5 Sox17 NM_001289466 chr1 - 4481008 4489935
> matchpeaksgenes -u 2500 H3K4me3_mESC.bed refGene.txt | gawk '{print $5}' | sort | uniq | head -10
0610005C13Rik
0610007P14Rik
0610009B22Rik
0610009L18Rik
0610009O20Rik
0610010B08Rik
0610010F05Rik
0610010K14Rik
0610012G03Rik
0610030E20Rik